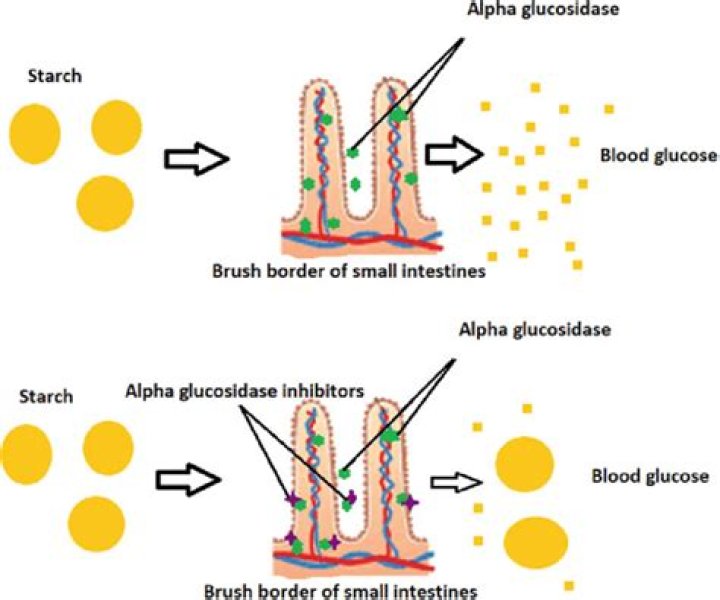
Acid alpha-glucosidase, also called α-1,4-glucosidase and acid maltase, is an enzyme (EC 3.2. 1.20) that helps to break down glycogen in the lysosome.
What are the symptoms of Pompe disease?
What are the symptoms of each type of Pompe disease?
- Weak muscles.
- Poor muscle tone.
- Enlarged liver.
- Failure to gain weight and grow at the expected rate (failure to thrive)
- Trouble breathing.
- Feeding problems.
- Infections in the respiratory system.
- Problems with hearing.
What enzyme is deficiency in Pompe disease?
Pompe disease is a rare (estimated at 1 in every 40,000 births), inherited and often fatal disorder that disables the heart and skeletal muscles. It is caused by mutations in a gene that makes an enzyme called acid alpha-glucosidase (GAA).
How does Pompe disease affect lysosomes?
Pompe disease is a lysosomal storage disorder in which acid alpha-glucosidase (GAA) is deficient or absent. Deficiency of this lysosomal enzyme results in progressive expansion of glycogen-filled lysosomes in multiple tissues, with cardiac and skeletal muscle being the most severely affected.
What are the lysosomal storage disorders and what are the symptoms?
Symptoms of Lysosomal Storage Diseases
- Delay in intellectual and physical development.
- Seizures.
- Facial and other bone deformities.
- Joint stiffness and pain.
- Difficulty breathing.
- Problems with vision and hearing.
- Anemia, nosebleeds, and easy bleeding or bruising.
- Swollen abdomen due to enlarged spleen or liver.
How does Fabry disease affect lysosomes?
Fabry disease is caused by a mutation in a gene called GLA, which provides instructions for making an enzyme known as alpha-galactosidase A. Lysosomes require this enzyme to properly break down large fat molecules inside the body’s cells.
What is the pathogenesis of alpha alpha glucosidase deficiency?
Pathogenesis and Etiology. The biochemical defect involves acid alpha-glucosidase (GAA; acid maltase) activity (Fig. 32.10 ). 171,179,180 This enzyme catalyzes the lysosomal pathway of glycogen degradation, presumably used during normal turnover of cellular constituents.
Is lysosomal GAA deficiency dominant or recessive?
Lysosomal GAA deficiency is an autosomal recessive disorder with considerable allelic heterogeneity. It is caused by mutations in the gene encoding lysosomal acid alpha-1,4-glucosidase (GAA), located at 17q25.2-q25.3 [8,9]. More than 550 mutations causing the disorder have been reported [10].
What are the treatment options for α-glucosidase deficiency?
Acid α-Glucosidase Deficiency. Patients with GAA deficiency may need intermittent or permanent mechanically assisted ventilation. 184 A regimen combining high-protein diet and aerobic exercise has improved strength and respiratory function in some patients 213 but needs to be validated in a large cohort.
What is the pathophysiology of lysosomal acidosis?
This autosomal recessive metabolic disorder is caused by mutations in lysosomal acid alpha-glucosidase (acid maltase), leading to pathologic intracellular accumulations of glycogen and exaggerated lysosomal function.