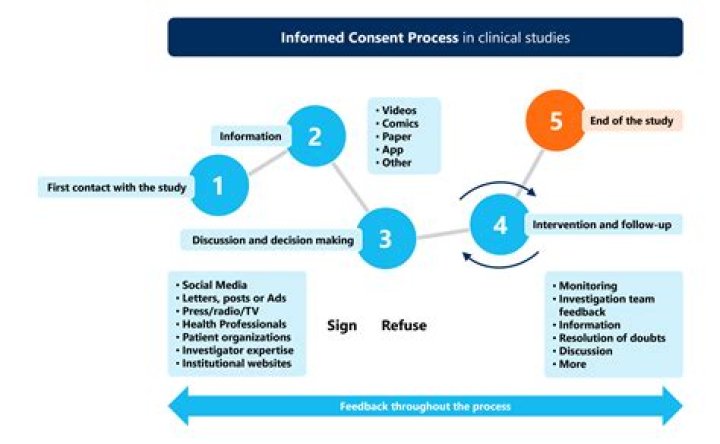
The Joint Commission requires documentation of all the elements of informed consent “in a form, progress notes or elsewhere in the record.” The following are the required elements for documentation of the informed consent discussion: (1) the nature of the procedure, (2) the risks and benefits and the procedure, (3) …
What steps should you take to ensure that the participants are able to provide informed consent?
The entire informed consent process involves giving a subject adequate information concerning the study, providing adequate opportunity for the subject to consider all options, responding to the subject’s questions, ensuring that the subject has comprehended this information, obtaining the subject’s voluntary agreement …
What must occur for your participants to give informed consent that maintains ethical guidelines?
NOTE: Voluntary informed consent means that the person involved should have legal capacity to give consent; should be so situated as to be able to exercise free power of choice, without the intervention of any element of force, fraud, deceit, duress, over reaching, or other ulterior form of constraints or coercion; and …
What is informed consent example?
I have read and I understand the provided information and have had the opportunity to ask questions. I understand that my participation is voluntary and that I am free to withdraw at any time, without giving a reason and without cost. I understand that I will be given a copy of this consent form.
What are the two types of informed consent?
Types of Informed Consent
- Implied consent: Implied consent refers to when a patient passively cooperates in a process without discussion or formal consent.
- Verbal consent: A verbal consent is where a patient states their consent to a procedure verbally but does not sign any written form.
What are the OHRP and FDA guidance documents for IRBs?
Both OHRP and FDA have a range of guidance documents [6] that cover a variety of topics that may be useful to IRB administrators, IRB chairpersons, and other institutional officials when preparing written procedures for the IRB.
Are there any regulations that require clinical investigators to report to IRB?
Are there any regulations that require clinical investigators to report to the IRB when a study has been completed? IRBs are required to function under written procedures. One of these procedural requirements [21 CFR 56.108 (a) (3)] requires ensuring “prompt reporting to the IRB of changes in a research activity.”
What documents are required to be submitted to the IRB?
A list of documents to be submitted to the IRB (e.g., protocol, informed consent form, investigator brochure, recruitment materials, HHS-approved protocol and sample informed consent form). Timelines for receipt of submissions, scheduling IRB review, and document distribution to IRB members.
What are the possible actions taken by the IRB?
The range of possible actions that can be taken by the IRB (e.g., approve, require modifications in to secure approval, disapprove, suspend or terminate approval of a study). 4. Conducting review via expedited review procedures. [9] A list of documents required for submission and provided to the person conducting the expedited review.